Element-specific anisotropic growth of shaped platinum alloy nanocrystals
 |
Lin Gan, Chunhua Cui, Marc Heggen, Fabio Dionigi, Stefan Rudi, Peter Strasser
Morphological shape in chemistry and biology owes its existence to anisotropic growth and is closely coupled to distinct functionality. Although much is known about the principal growth mechanisms of monometallic shaped nanocrystals, the anisotropic growth of shaped alloy nanocrystals is still poorly understood. Using aberration-corrected scanning transmission electron microscopy, we reveal an element-specific anisotropic growth mechanism of platinum (Pt) bimetallic nano-octahedra where compositional anisotropy couples to geometric anisotropy.
A Pt-rich phase evolves into precursor nanohexapods, followed by a slower step-induced deposition of an M-rich (M = Ni, Co, etc.) phase at the concave hexapod surface forming the octahedral facets. Our finding explains earlier reports on unusual compositional segregations and chemical degradation pathways of bimetallic polyhedral catalysts and may aid rational synthesis of shaped alloy catalysts with desired compositional patterns and properties.
Science 2014, 346, 1502-1506 | DOI: 10.1126/science.1261212
Resonance Raman Spectroscopy on [NiFe] Hydrogenase Provides Structural Insights into Catalytic Intermediates and Reactions
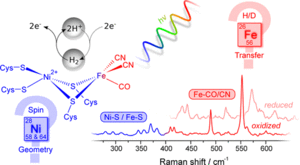
Marius Horch, Janna Schoknecht, Maria Andrea Mroginski, Oliver Lenz, Peter Hildebrandt, and Ingo Zebger
[NiFe] hydrogenases catalyze the reversible cleavage of hydrogen and, thus, represent model systems for the investigation and exploitation of emission-free energy conversion processes. Valuable information on the underlying molecular mechanisms can be obtained by spectroscopic techniques that monitor individual catalytic intermediates.
Here, we employed resonance Raman spectroscopy and extended it to the entire binuclear active site of an oxygen-tolerant [NiFe] hydrogenase by probing the metal–ligand modes of both the Fe and, for the first time, the Ni ion. Supported by theoretical methods, this approach allowed for monitoring H-transfer from the active site and revealed novel insights into the so far unknown structure and electronic configuration of the hydrogen-binding intermediate of the catalytic cycle, thereby providing key information about catalytic intermediates and reactions of biological hydrogen activation.
J. Am. Chem. Soc. 2014, 136, 9870−9873 | DOI: 10.1021/ja505119q
Sol–gel method for synthesis of Mn–Na2WO4/SiO2 catalyst for methane oxidative coupling
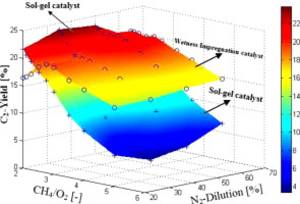
H.R. Godini, A. Gili, O. Görke, S. Arndt, U. Simon, A. Thomas, R. Schomäcker, G. Wozny
In this experimental study, a novel sol–gel method was developed to synthesize a 1.9%Mn–5%Na2WO4/SiO2 catalyst for oxidative coupling of methane (OCM) reactions. The performance of the synthesized catalyst was investigated in fixed-bed and porous packed-bed membrane reactors.
Particularly, the effects of operating temperature, methane-to-oxygen ratio and nitrogen dilution on the performance of this catalyst were investigated. It was observed that for high values of methane conversion, the sol–gel Mn–Na2WO4/SiO2 catalyst provides 5–15% higher selectivity toward the desired products (C2: C2H4 + C2H6) than the Mn–Na2WO4/SiO2 catalyst prepared by the incipient wetness impregnation method.
It was also observed that for a similar set of experiments, the C2-selectivity of the sol–gel catalyst is affected relatively less by the variation of methane-to-oxygen ratio. As a result, this catalyst can be exploited under the low methane-to-oxygen feed ratio which provides an efficient performance in both the OCM reactor and the OCM process scale. The best observed performance of the sol–gel catalyst in the packed-bed membrane reactor is 78% C2-selectivity, 64% ethylene-selectivity and 24.2% C2-yield under 20% nitrogen dilution.
Catalysis Today 2014, 236, 12−22 | DOI: 10.1016/j.cattod.2014.01.005
Native-like Photosystem II Superstructure at 2.44 A Resolution through Detergent Extraction from the Protein Crystal
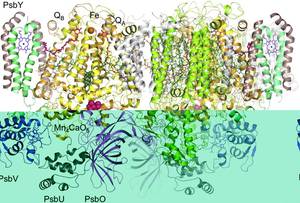
J. Hellmich, M. Bommer, A. Burkhardt, M. Ibrahim, J. Kern, A. Meents, F. Müh, H. Dobbek, A. Zouni
Photosystem II (PSII) catalyzes a key step in photosynthesis, the oxidation of water to oxygen. Excellent structural models exist for the dimeric PSII core complex of cyanobacteria, but higher order physiological assemblies readily dissociate when solubilized from the native thylakoid membrane with detergent.
Here, we describe the crystallization of PSII from Thermosynechococcus elongatus with a postcrystallization treatment involving extraction of the detergent C12E8. This resulted in a transition from Type II to Type I-like membrane protein crystals and improved diffraction to 2.44 Å resolution.
The obtained PSII packing in precise rows, interconnected by specific pairs of galactolipids and a loop in the PsbO subunit specific to cyanobacteria, is superimposable with previous electron microscopy images of the thylakoid membrane. The study provides a detailed model of such a superstructure and its organization of light-harvesting pigments with possible implications for the understanding of their efficient use of solar energy.
Structure 2014, 22, 1607−1615 | DOI: 10.1016/j.str.2014.09.007
Published online 30 October 2014
Enhanced catalytic performance of MnxOy–Na2WO4/SiO2 for the oxidative coupling of methane using an ordered mesoporous silica support
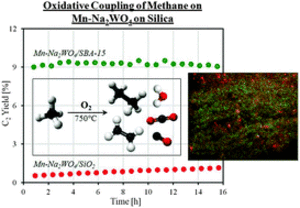
M. Yildid, Y. Aksu, U. Simon, K. Kailasam, O. Goerke, F. Rosowski, R. Schomäcker, A. Thomas, S. ArndtZouni, Athina
The oxidative coupling of methane is a highly promising reaction for its direct conversion. Silica supported MnxOy–Na2WO4 is a suitable catalyst for this reaction. In this study, a variety of different SiO2 materials have been tested as supports. Surprisingly, the application of ordered mesoporous silicas, here exemplarily shown for SBA-15 as support materials, greatly enhances the catalytic performance. The CH4 conversion increased two fold and also the C2 selectivity is strongly increased.
Chemical Communications 2014, 50, 14440−14442| DOI: 10.1039/C4CC06561A
Published online 10 October 2014
How to energize an electron
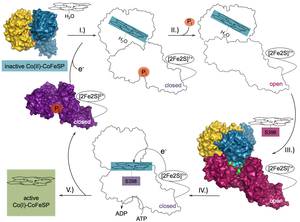
Sandra E. Hennig, Sebastian Goetzl, Jae-Hun Jeoung, Martin Bommer, Friedhelm Lendzian, Peter Hildebrandt, Holger Dobbek
Thermodynamically unfavourable electron transfers are enabled by coupling to an energy-supplying reaction. How the energy is transduced from the exergonic to the endergonic process is largely unknown. Here we provide the structural basis for an energy transduction process in the reductive activation of B12-dependent methyltransferases. The transfer of one electron from an activating enzyme to the cobalamin cofactor is energetically uphill and relies on coupling to an ATPase reaction.
Our results demonstrate that the key to coupling is, besides the oxidation state-dependent complex formation, the conformational gating of the electron transfer. Complex formation induces a substitution of the ligand at the electron-accepting Co ion. Addition of ATP initiates electron transfer by provoking conformational changes that destabilize the complex. We show how remodelling of the electron-accepting Co2+ promotes ATP-dependent electron transfer; an efficient strategy not seen in other electron-transferring ATPases.
Nature Communications | 5:4626 | DOI: 10.1038/ncomms5626
Published online 11 August 2014
Taking snapshots of photosynthetic water oxidation using femtosecond X-ray diffraction and spectroscopy
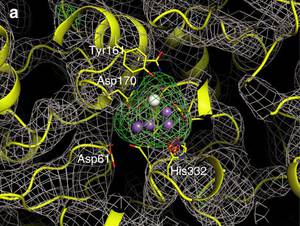 |
Jan Kern, Rosalie Tran, Roberto Alonso-Mori, Sergey Koroidov, Nathaniel Echols, Johan Hattne, Mohamed Ibrahim, Sheraz Gul, Hartawan Laksmono, Raymond G. Sierra, Richard J. Gildea, Guangye Han, Julia Hellmich, Benedikt Lassalle-Kaiser, Ruchira Chatterjee, Aaron S. Brewster, Claudiu A. Stan, Carina Glöckner, Alyssa Lampe, Dörte DiFiore, Despina Milathianaki, Alan R. Fry, M. Marvin Seibert, Jason E. Koglin, Erik Gallo, Jens Uhlig, Dimosthenis Sokaras, Tsu-Chien Weng, Petrus H. Zwart, David E. Skinner, Michael J. Bogan, Marc Messerschmidt, Pieter Glatzel, Garth J. Williams, Sébastien Boutet, Paul D. Adams, Athina Zouni, Johannes Messinger, Nicholas K. Sauter, Uwe Bergmann, Junko Yano, Vittal K. Yachandra
The dioxygen we breathe is formed by light-induced oxidation of water in photosystem II. O2 formation takes place at a catalytic manganese cluster within milliseconds after the photosystem II reaction centre is excited by three single-turnover flashes. Here we present combined X-ray emission spectra and diffraction data of 2-flash (2F) and 3-flash (3F) photosystem II samples, and of a transient 3F’ state (250 μs after the third flash), collected under functional conditions using an X-ray free electron laser. This study represents the initial frames in a molecular movie of the structural changes during the catalytic reaction in photosystem II.
Nature Communications | 5:4371 | DOI: 10.1038/ncomms5371
Published online 09 July 2014
A Molecular Approach to Self-Supported Cobalt-Substituted ZnO Materials as Remarkably Stable Electrocatalysts for Water Oxidation
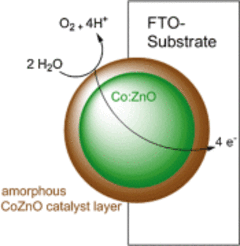
Johannes Pfrommer, Michael Lublow, Anahita Azarpira, Caren Göbel, Marcel Lücke, Alexander Steigert, Martin Pogrzeba, Prashanth W. Menezes, Anna Fischer, Thomas Schedel-Niedrig, and Matthias Driess
In regard to earth-abundant cobalt water oxidation catalysts, very recent findings show the reorganization of the materials to amorphous active phases under catalytic conditions. To further understand this concept, a unique cobalt-substituted crystalline zinc oxide (Co:ZnO) precatalyst has been synthesized by low-temperature solvolysis of molecular heterobimetallic Co4−xZnxO4 (x=1–3) precursors in benzylamine.
Its electrophoretic deposition onto fluorinated tin oxide electrodes leads after oxidative conditioning to an amorphous self-supported water-oxidation electrocatalyst, which was observed by HR-TEM on FIB lamellas of the EPD layers. The Co-rich hydroxide-oxidic electrocatalyst performs at very low overpotentials (512 mV at pH 7; 330 mV at pH 12), while chronoamperometry shows a stable catalytic current over several hours.
This paper has been selected as a hot paper in Angewandte Chemie.
Angew. Chem. Int. Ed. 2014; DOI: 10.1002/anie.201400243
Published online 28 April 2014.
Sites for Methane Activation on Lithium-Doped Magnesium Oxide Surfaces
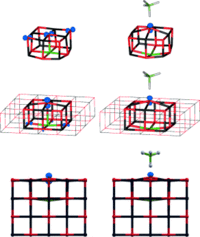 |
Karolina Kwapien, Joachim Paier, Joachim Sauer, Michael Geske, Ulyana Zavyalova, Raimund Horn, Pierre Schwach, Annette Trunschke, and Robert Schlögl
Density functional calculations yield energy barriers for H abstraction by oxygen radical sites in Li-doped MgO that are much smaller (12±6 kJ mol−1) than the barriers inferred from different experimental studies (80–160 kJ mol−1). This raises further doubts that the Li+O.− site is the active site as postulated by Lunsford. From temperature-programmed oxidative coupling reactions of methane (OCM), we conclude that the same sites are responsible for the activation of CH4 on both Li-doped MgO and pure MgO catalysts.
Angew. Chem. Int. Ed. 2014; DOI: 10.1002/anie.201310632
Published online on 23 April 2014.
Reversible [4Fe-3S] cluster morphing in an oxygen tolerant [NiFe] hydrogenase
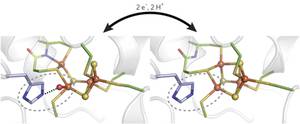
Stefan Frielingsdorf, Johannes Fritsch, Andrea Schmidt, Mathias Hammer, Julia Löwenstein, Elisabeth Siebert, Vladimir Pelmenschikov, Tina Jaenicke, Jacqueline Kalms, Yvonne Rippers, Friedhelm Lendzian, Ingo Zebger, Christian Teutloff, Martin Kaupp, Robert Bittl, Peter Hildebrandt, Bärbel Friedrich, Oliver Lenz & Patrick Scheerer
Certain oxygen-tolerant hydrogenases contain a unique [4Fe-3S] cluster near the catalytic site, but the role of this cofactor is not fully understood. Crystallographic, spectroscopic and computational data now provide evidence for redox-dependent transformations of this cluster, potentially explaining how specialized hydrogenases can safely reduce inhibitory O2.
Nat. Chem. Biol. 2014; DOI: 10.1038/nchembio.1500
Published online on 6 April 2014.
Rubredoxin-related Maturation Factor Guarantees Metal Cofactor Integrity during Aerobic Biosynthesis of Membrane-bound [NiFe] Hydrogenase
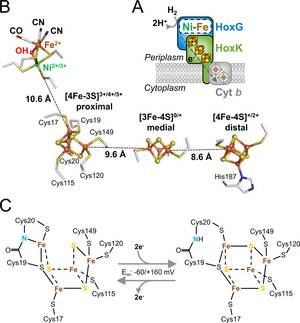
J. Fritsch, E. Siebert, J. Priebe, I. Zebger, F. Lendzian, C. Teutloff, B. Friedrich, O. Lenz
The membrane-bound [NiFe] hydrogenase (MBH) supports growth of Ralstonia eutropha H16 with H2 as the sole energy source. The enzyme undergoes a complex biosynthesis process that proceeds during cell growth even at ambient O2 levels and involves 14 specific maturation proteins. One of these is a rubredoxin-like protein, which is essential for biosynthesis of active MBH at high oxygen concentrations but dispensable under microaerobic growth conditions.
To obtain insights into the function of HoxR, we investigated the MBH protein purified from the cytoplasmic membrane of hoxR mutant cells. Compared with wild-type MBH, the mutant enzyme displayed severely decreased hydrogenase activity. Electron paramagnetic resonance and infrared spectroscopic analyses revealed features resembling those of O2-sensitive [NiFe] hydrogenases and/or oxidatively damaged protein. The catalytic center resided partially in an inactive Niu-A-like state, and the electron transfer chain consisting of three different Fe-S clusters showed marked alterations compared with wild-type enzyme. Purification of HoxR protein from its original host, R. eutropha, revealed only low protein amounts. Therefore, recombinant HoxR protein was isolated from Escherichia coli.
Unlike common rubredoxins, the HoxR protein was colorless, rather unstable, and essentially metal-free. Conversion of the atypical iron-binding motif into a canonical one through genetic engineering led to a stable reddish rubredoxin. Remarkably, the modified HoxR protein did not support MBH-dependent growth at high O2. Analysis of MBH-associated protein complexes points toward a specific interaction of HoxR with the Fe-S cluster-bearing small subunit. This supports the previously made notion that HoxR avoids oxidative damage of the metal centers of the MBH, in particular the unprecedented Cys6[4Fe-3S] cluster.
The Journal of Biological Chemistry 2014, 289, 7982-7993 | DOI: 10.1074/jbc.M113.544668
Published online on 1 January 2014.
Redox Non-Innocence of a N-Heterocyclic Nitrenium Cation Bound to a Nickel–Cyclam Core
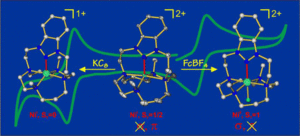
Florian Heims, Florian Felix Pfaff, Sarah-Luise Abram, Erik R. Farquhar, Maurizio Bruschi, Claudio Greco, and Kallol Ray
The redox properties of Ni complexes bound to a new ligand, [DMC-nit]+, where a N-heterocyclic nitrenium group is anchored on a 1,4,8,11-tetraazacyclotetradecane backbone, have been examined using spectroscopic and DFT methods. Ligand-based [(DMC-nit)Ni]2+/+ reduction and metal-based [(DMC-nit)Ni]2+/3+ oxidation processes have been established for the [(DMC-nit)Ni]+/2+/3+ redox series, which represents the first examples of nitrenium nitrogen (Nnit)-bound first-row transition-metal complexes.
An unprecedented bent binding mode of Nnit in [(DMC-nit)Ni]2+ is observed, which possibly results from the absence of any Nnit→Ni σ-donation. For the corresponding [(DMC-nit)Ni(F)]2+ complex, σ-donation is dominant, and hence a coplanar arrangement at Nnit is predicted by DFT. The binding of the triazolium ion to Ni enables new chemistry (formate oxidation) that is not observed in a derivative that lacks this functional group. Thus the N-heterocyclic nitrenium ligand is a potentially useful and versatile reagent in transition-metal-based catalysis.
J. Am. Chem. Soc., 2014, 136, 582–585; DOI: 10.1021/ja4099559
Published online 20 December 2013.