O-O Bond activation in heterobimetallic peroxides: synthesis of the peroxide [LNi(μ,η2:η2-O2)K] and its conversion into a bis(μ-hydroxo) nickel zinc complex
 |
Shenglai Yao, Yun Xiong, Matthias Vogt, Hansjörg Grützmacher, Christian Herwig, Christian Limberg, and Matthias Driess
Angew. Chem. Int. Ed. 2009, 48, 8107;
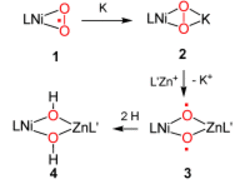
O![]() O bond activation can be achieved depending on the nature of the heterometal M in [LNi(μ,η2:η2-O2)M] complexes. The reduction of the nickel(II) superoxide 1 with potassium affords the peroxide 2, which, upon replacement of the K+ ion in 2 by the non-redox-active L′Zn+ ion, leads to transient 3, which subsequently abstracts two solvent hydrogen atoms to give the heterobimetallic complex 4. L, L′=β-diketiminates.
O bond activation can be achieved depending on the nature of the heterometal M in [LNi(μ,η2:η2-O2)M] complexes. The reduction of the nickel(II) superoxide 1 with potassium affords the peroxide 2, which, upon replacement of the K+ ion in 2 by the non-redox-active L′Zn+ ion, leads to transient 3, which subsequently abstracts two solvent hydrogen atoms to give the heterobimetallic complex 4. L, L′=β-diketiminates.
A Redox Non-Innocent Ligand Controls the Life Time of a Reactive Quartet Excited State - An MCSCF Study of [Ni(H)(OH)]+
 |
Yavuz Dede, Xinhao Zhang, Maria Schlangen, Helmut Schwarz, and Mu-Hyun Baik
J. Am. Chem. Soc. 2010, 131, 12634;
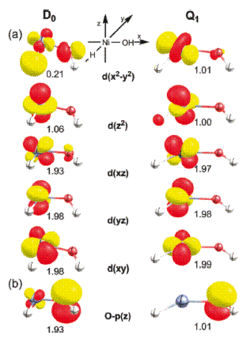
The electronic structures of the low and high-spin states of the cationic complex [Ni(H)(OH)]+ that was previously found to be highly reactive toward CH4 and O2 were examined. Earlier computational work suggested that the low-spin doublet state D0 of the NiIII-d7 system is significantly lower in energy than its high-spin quartet analogue Q1. Recent DFT-studies indicated, however, that Q1 is the reactive species requiring Q1 to have a sufficiently long lifetime for undergoing thermal reactions with the small molecule reactants under single collision conditions in the gas phase.
These observations raise the question as to why Q1 does not spontaneously undergo intersystem crossing. Our work based on DFT, coupled-cluster and MCSCF calculations suggests that the hydroxyl ligand behaves as a redox noninnocent ligand and becomes oxidized to formally afford an electronic structure that is consistent with a NiII-(OH)• species. As a result, the doublet and quartet ground states are not related by a single electron spin flip and the intersystem crossing becomes inhibited, as indicated by unexpectedly small spin-orbit coupling constants.
After extensive sampling of the potential energy surfaces, we concluded that there is no direct way of converting Q1 to the ground state doublet D0. Alternative multistep pathways for the Q1 --> D0 decay involving doublet excited states were also evaluated and found to be energetically not accessible under the experimental conditions.
Cyanobacterial photosystem II at 2.9-Å resolution and the role of quinones, lipids, channels and chloride
 |
Albert Guskov, Jan Kern, Azat Gabdulkhakov, Matthias Broser, Athina Zouni & Wolfram Saenger
Nat. Struct. Mol. Biol. 2009,
16, 334 -342
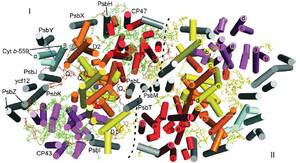
Photosystem II (PSII) is a large homodimeric protein–cofactor complex located in the photosynthetic thylakoid membrane that acts as light-driven water:plastoquinone oxidoreductase. The crystal structure of PSII from Thermosynechococcus elongatus at 2.9-Å resolution allowed the unambiguous assignment of all 20 protein subunits and complete modeling of all 35 chlorophyll a molecules and 12 carotenoid molecules, 25 integral lipids and 1 chloride ion per monomer.
The presence of a third plastoquinone QC and a second plastoquinone-transfer channel, which were not observed before, suggests mechanisms for plastoquinolplastoquinone exchange, and we calculated other possible water or dioxygen and proton channels. Putative oxygen positions obtained from a Xenon derivative indicate a role for lipids in oxygen diffusion to the cytoplasmic side of PSII. The chloride position suggests a role in proton-transfer reactions because it is bound through a putative water molecule to the Mn4Ca cluster at a distance of 6.5 Å and is close to two possible proton channels.
Molecular Basis for the Electric Field Modulation of Cytochrome c Structure and Function
 |
Pablo M. De Biase, Damián Alvarez Paggi, Fabio Doctorovich, Peter Hildebrandt, Dario A. Estrin, Daniel H. Murgida, and Marcelo A. Marti
JACS 131 (2009), 16248 - 16256.
Cytochrome c (Cyt) is a small soluble heme protein with a hexacoordinated heme and functions as an electron shuttle in the mitochondria and in early events of apoptosis when released to the cytoplasm. Using molecular dynamics simulations, we show here that biologically relevant electric fields induce an increased mobility and structural distortion of key protein segments that leads to the detachment of the sixth axial ligand Met80 from the heme iron.
This electric-field-induced conformational transition is energetically and entropically driven and leads to a pentacoordinated high spin heme that is characterized by a drastically lowered reduction potential as well as by an increased peroxidase activity. The simulations provide a detailed atomistic picture of the structural effects of the electric field on the structure of Cyt, which allows a sound interpretation of recent experimental results. The observed conformational change may modulate the electron transfer reactions of Cyt in the mitochondria and, furthermore, may constitute a switch from the redox function in the respiratory chain to the peroxidase function in the early events of apoptosis.
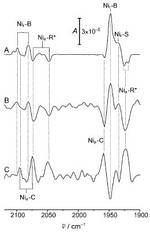
Monitoring Catalysis of the Membrane-Bound Hydrogenase from Ralstonia eutropha H16 by Surface-Enhanced IR Absorption Spectroscopy
 |
Nattawadee Wisitruangsakul, Oliver Lenz, Marcus Ludwig, Bärbel Friedrich, Friedhelm Lendzian, Peter Hildebrandt, and Ingo Zebger
Ang. Chem. Int. Ed. 2009, 48, 611-613
In the past years, there is a strongly growing interest in hydrogenases since these enzymes may catalyse either hydrogen splitting or hydrogen formation, i.e., reactions that are of technological importance for energy storage and conversion.
To utilize enzymes for catalyzing these reactions is a challenge since it requires immobilization of the proteins on electrically conducting materials under preservation of the native structure and function. It is, therefore, desirable to develop a methodology of sufficient sensitivity and selectivity that allows probing the active site structure of immobilized enzyme during the catalytic process. In this work we have shown that surface enhanced infrared absorption (SEIRA) spectroscopy fulfils these requirements.
Moreover, the results demonstrate that the membrane-bound hydrogenase from Ralstonia eutropha, retains its structural and functional integrity in the
immobilized state such that the biocatalytic process can be monitored in situ by SEIRA spectroscopy. The findings are of particular importance in view of the remarkable oxygen-tolerance of this enzyme which makes it a highly promising candidate for biotechnological applications.
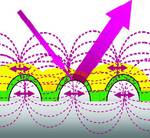
Novel Au-Ag Hybrid Device for Electrochemical SE(R)R Spectroscopy in a Wide Potential and Spectral Range
Jiu-Ju Feng, Ulrich Gernert, Murat Sezer, Uwe Kuhlmann, Daniel H. Murgida, Christin David, Marten Richter, Andreas Knorr, Peter Hildebrandt, and Inez M. Weidinger
Nano Letters 2009, 9, 298 - 303
A nanostructured gold-silver-hybrid electrode for SER spectroelectrochemistry was developed which advantageously combines the electrochemical properties and chemical stability of Au and the strong surface enhancement of (resonance) Raman scattering by Ag.
The layered device consists of a massive nanoscopically rough Ag electrode, a thin (2 nm) organic layer, and a ca. 20 nm thick Au film that may be coated by self-assembled monolayers for protein adsorption. The SERR-spectroscopic and electrochemical performance of this device is demonstrated using the heme protein cytochrome c as a benchmark model system, thereby extending, for the first time, SE(R)R studies of molecules on Au surfaces to excitation in the violet spectral range.
The enhancement factor is only slightly lower than for Ag electrodes which can be rationalized in terms of an efficient transfer of plasmon resonance excitation from the Ag to the Au coating. This mechanism, which requires a thin dielectric layer between the two metals, is supported by theoretical calculations.
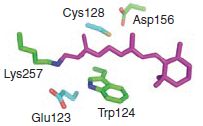
Bi-stable neural state switches
André Berndt, Ofer Yizhar, Lisa A Gunaydin, Peter Hegemann & Karl Deisseroth
Nature Neuroscience 2009, 12, 229 - 234
Here we describe bi-stable channelrhodopsins that convert a briefpulse of light into a stable step in membrane potential. Thesemolecularly engineered probes nevertheless retain millisecondscaletemporal precision. Photocurrents can be precisely initiatedand terminated with different colors of light, but operate atvastly longer time scales than conventional channelrhodopsinsas a result of modification at the C128 position that extends thelifetime of the open state.
Because of their enhanced kineticstability, these step-function tools are also effectively responsiveto light at orders of magnitude lower intensity than wild-typechannelrhodopsins. These molecules therefore offer importantnew capabilities for a broad range of in vivo applications.